
随着医药行业的快速发展和监管要求的不断提高,药企实验室在保障产品质量、推动研发创新方面发挥着关键作用。而信息化系统的应用,不仅提升了实验室管理的效率和准确性,更成为药企实现法规符合性的重要手段。本文旨在浅析药企实验室信息化系统与法规符合性策略,探讨如何通过信息化手段,确保实验室管理符合相关法规要求,提升药企的整体合规水平。
随着医药行业的快速发展和监管要求的不断提高,药企实验室在保障产品质量、推动研发创新方面发挥着关键作用。而信息化系统的应用,不仅提升了实验室管理的效率和准确性,更成为药企实现法规符合性的重要手段。本文旨在浅析药企实验室信息化系统与法规符合性策略,探讨如何通过信息化手段,确保实验室管理符合相关法规要求,提升药企的整体合规水平。
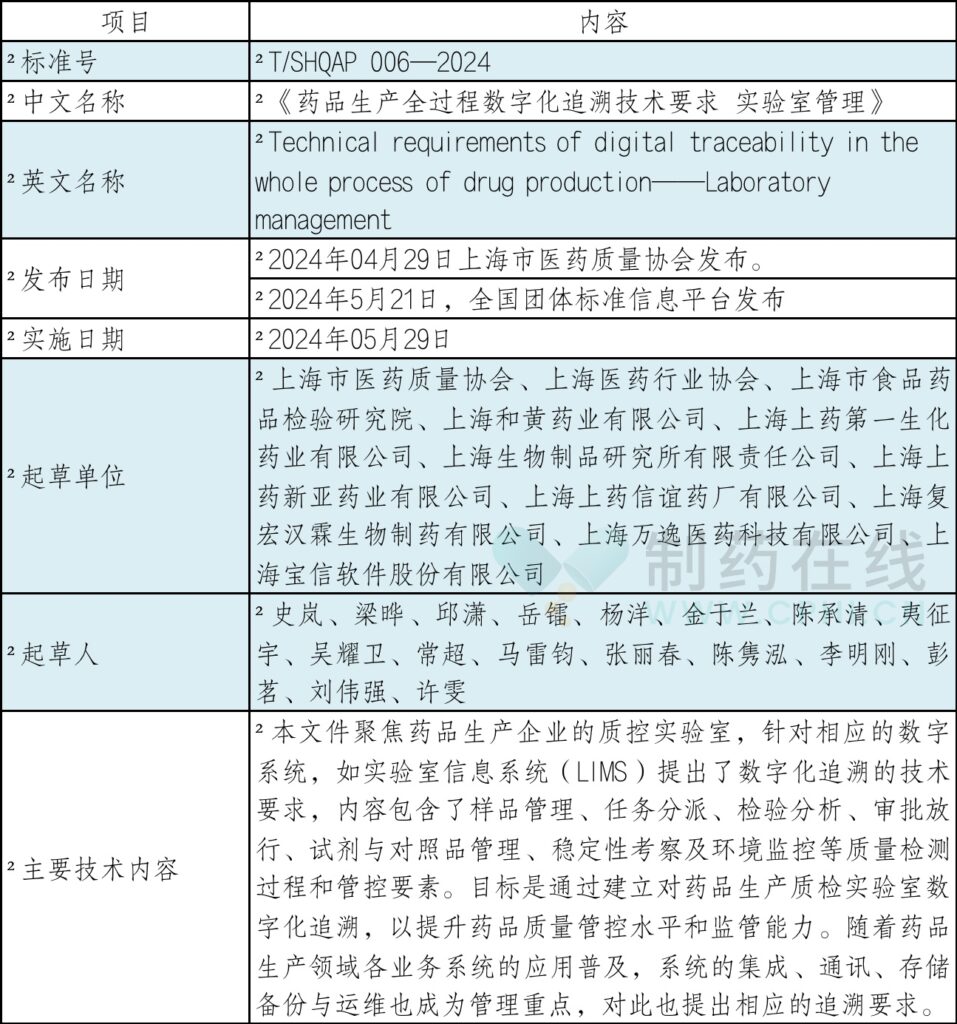
2024年5月21日,全国团体标准信息平台正式发布了上海市医药质量协会组织起草的T/SHQAP 006—2024《药品生产全过程数字化追溯技术要求 实验室管理》标准,该标准将于2024年5月29日起正式实施。此标准的出台标志着上海市在药品生产数字化追溯领域迈出了重要一步,尤其是在实验室信息管理方面,为行业树立了新的标杆,在药品生产全过程数字化追溯体系技术要求–实验室信息管理标准率先提出了上海方案。
一、药企实验室信息化系统法规符合性
随着计算机化系统在药品生产和质量管理中的全面渗透,GMP文件系统中的核心环节——GMP记录及其管理,正面临深刻的变革。为确保企业GMP的有效实施,关键在于如何充分利用计算机化系统的特点,优化GMP记录的管理。
电子记录,作为依托计算机系统进行创建、修改、维护、存档、检索及发送的文本、图表、数据、声音、图像等电子形式信息的集合,相较于传统纸介质记录,展现了显著的优势。它不仅便于数据的统计分析,还作为质量追溯、质量系统分析和持续改进的关键依据,其检索的便捷性和数据采集的完整性更是传统记录无法比拟的。因此,构建和完善计算机化系统下的GMP电子记录管理,对于提升药品生产和质量管理的效率与水平具有重要意义。
笔者结合国家及各地方GMP监管机构的检查缺陷案例,梳理了GMP符合性检查中常见的实验室管理数据可靠性典型缺陷案例,示例如下:
案例1:样品追踪迷踪
在某次GMP检查中,发现某药企QC实验室的样品登记台账如同未完待续的故事书,关键章节缺失——样品取样记录竟无踪影。这不仅让样品的身份成了谜,更让数据的追溯链断裂,无法确保每份样品的测试结果都能找到其原始的“出生证明”。
案例2:时间旅行的报告
测试时间与记录报告上的时间不符,经调查发现是系统时间被人为修改。这种“时间旅行”的行为,严重破坏了数据的真实性和时效性。
案例3:HPLC的灰色地带
HPLC(高效液相色谱仪)的积分参数被悄然调整,系统适用性遭受质疑。更令人担忧的是,缺乏审计追踪功能或该功能未激活,使得对数据的任何修改都如同在黑暗中行走,无法留下清晰的足迹。
案例4:图谱的“分身术”
复制粘贴的陷阱:同一张图谱被用于不同批次产品的测试报告,这种数据复用的行为,如同变魔术般欺骗了审核者的眼睛,严重违反了数据独立性的原则。
案例5:数据的消失与伪装
数据黑洞:数据被删除或覆盖,且未给出合理解释。这种数据的“蒸发”和“伪装”,如同在数据海洋中投下了迷雾弹,让真相变得扑朔迷离。
案例6:数据迁移的陷阱
原始数据的失落:在数据转移过程中,原始数据如同被遗忘的行李,未被妥善保留。这种疏忽,让数据的追溯性和完整性受到了严重损害。
案例7:权限迷宫
计算机化系统内的权限和密码管理形同虚设,仿佛一扇未上锁的大门,任何人都能自由进出数据的世界。这种无序状态,为数据的篡改和误操作提供了温床,严重威胁着数据的准确性和安全性。
案例8:天平的沉默秘密
一台配备先进打印功能的天平,本应是数据记录的忠实守护者,却意外地保持了沉默。称量操作后,本应自动打印的记录未现身,且这些关键数据也未被妥善存档。
案例9:预演的秘密
测试过程中,小试和试样的频繁出现,以及未加标注的预测试,如同实验前的彩排,却未遵循正式演出的规则。这不仅扰乱了数据的纯净性,还可能掩盖了实验过程中潜在的问题。
案例10:溶液配置的“黑箱”
配置盲区:标准溶液、缓冲溶液等关键试剂的配置过程如同黑箱操作,缺乏必要的配置记录和信息。这种不透明性,让试剂的质量和稳定性充满了不确定性。
2010年版GMP第一百六十三条规定,如使用电子数据处理系统、照相技术或其他可靠方式记录数据资料,应当有所用系统的操作规程;记录的准确性应当经过核对。使用电子数据处理系统的,只有经授权的人员方可输入或更改数据,更改和删除情况应当有记录;应当使用密码或其他方式来控制系统的登录;关键数据输入后,应当由他人独立进行复核。用电子方法保存的批记录,应当采用磁带、缩微胶卷、纸质副本或其他方法进行备份,以确保记录的安全,且数据资料在保存期内便于查阅。
《药品记录与数据管理要求(试行)》的第四章对电子记录管理提出了详细要求。首先,采用电子记录的计算机(化)系统需满足一系列设施与配置,包括安装在适当位置、配备稳定安全的网络环境和可靠的信息安全平台、实现信息传输和数据共享等。其次,系统应保证记录时间与系统时间的真实性、准确性,能显示并打印电子记录数据,并需定期备份数据,确保在变更或升级时数据可查阅与追溯。此外,电子记录需实现操作权限与用户登录管理,确保登录用户的唯一性与可追溯性,并遵循电子签名法相关规定。最后,系统验证项目需根据系统基础架构、功能与业务功能确定验证范围与程度,确保系统功能符合预定用途。这些要求旨在确保药品记录与数据的真实性、准确性和可追溯性。
二、药企实验室信息化系统建设策略
T/SHQAP 006—2024《药品生产全过程数字化追溯技术要求 实验室管理》标准结合药品行业的特性,详细规定了药品生产企业质量控制实验室在信息管理数字化追溯方面的通用要求。这些要求涵盖了追溯内容的明确性、索引的规范性、数据通讯的安全有效性、数据存储备份的可靠性以及运行维护的持续性等多个方面。通过这一系列规定,标准旨在确保药品生产全过程中实验室信息的准确性、可追溯性和安全性。
此项标准的发布与实施,对于上海市药品生产企业来说,不仅提供了明确的操作指导,还为其质量控制实验室的数字化追溯体系建设提供了有力支撑。该标准在药品生产全过程数字化追溯体系技术要求中,特别是在实验室信息管理方面,率先提出了具有上海特色的解决方案,为行业的持续发展注入了新的活力。
最新发布的《药品生产全过程数字化追溯技术要求 实验室管理》为药品实验室信息管理提供了全方位的指导。从样品检测流管理到实验记录管理,再到检验标准及方法管理,每一项都关乎药品质量的把控与追溯。
在初期实施阶段,我们建议优先部署核心的样品检测管理模块,确保检测流程的顺畅与准确。同时,库存试剂管理、仪器设备管理等基础功能也需同步上线,为实验室的稳定运行提供坚实保障。初期可以选择部分类型的检测样品进行试运营,以验证系统的实际效能。
随着系统的稳定运行与经验的积累,我们可以逐步扩展业务范围,如稳定性管理等,以进一步提升实验室的管理水平。此外,电子实验记录本模块的引入,将极大提升实验数据的记录与追溯效率,为药品质量的持续改进提供有力支持。
目前,实验室信息管理系统(LIMS)已融入前沿的计算机网络技术、数据库技术,并采纳了标准化的实验室管理理念,构建出一个全面且规范化的管理平台。该系统通过集成数据收集、处理、深入分析及质量控制等核心功能,以及提供法规遵从性工具,助力药企确保合规运营。该系统实现了对人员、设备、物料、方法、环境及检测的全方位智能管理,不仅推动了实验室业务流程的标准化和自动化,更在强化实验室安全管理、提升检测效率、发掘数据深层价值、优化资源配置及降低检测成本等方面展现出显著优势。
三、《药品生产全过程数字化追溯技术要求 实验室管理》标准信息
参考文献:
1、上海市医药质量协会、全国团体标准信息平台等